
Funded NYR-DRC Projects
New York Regional Diabetes Research Center
2024
Maria Jimenez-Gonzalez, PhD - MSSM
"Determining the role of pancreatic parasympathetic innervation in Type 1 Diabetes"
Type 1 Diabetes (T1D) is characterized by the autoimmune destruction of insulin-producing beta cells leading to hyperglycemia. In parallel, dysfunctional glucagon-producing alpha cells do not respond adequately to low glucose resulting in frequent episodes of hypoglycemia in T1D. Glucose regulation by the endocrine pancreas is highly regulated by the autonomic nervous system. Specifically, sympathetic nervous system activity inhibits insulin secretion and activates glucagon secretion, and leads to intra-islet vasoconstriction that together increase blood glucose. Importantly, sympathetic activity has been reported to exacerbate the early inflammatory process of several autoimmune diseases such as rheumatoid arthritis. However, the contributions of sympathetic innervation to metabolic and autoimmune pathology in T1D is largely unexplored. Existing structural and functional studies of sympathetic innervation in T1D are conflicting, in part because many of these studies used techniques that are not pancreas-specific. Importantly, there are no detailed longitudinal studies of the structure or function of pancreatic sympathetic innervation during both development of T1D and at later stages of the disease. Our previous studies and preliminary results strongly indicate increased islet sympathetic innervation occurs in early T1D, potentially contributing to accelerated immune activity, beta cell loss, hyperglycemia and hyperglucagonemia. Additionally, our data suggest that systemic and pancreas-specific sympathetic inactivation delays the onset of hyperglycemia in a mouse model of T1D. These data strongly support the hypothesis that increased sympathetic innervation accelerates immune destruction and hyperglycemia in T1D. We will test this hypothesis in two Specific Aims: 1) To determine the extent, changes and time-course of pancreatic islet sympathetic innervation in the NOD mouse model of T1D using unbiased 3D imaging; and, 2) Determining the effects of highly targeted ablation and activation of pancreatic sympathetic innervation in vivo on immune islet infiltration and metabolic function
Leigh Goedeke, PhD - MSSM
"Cobalamin-Dependent Metabolism and Steatotic Liver Disease"
Recent findings underscore the ability of common metabolites to direct cell function and fate. Yet, our understanding of mitochondrial biology and signaling requires deeper knowledge of the physiological mechanisms in human health and disease. This is particularly true today, as rising obesity and diabetes rates are exacerbating the existing crisis of steatotic liver disease and it’s end-stage complications. One metabolite that has recently gained attention as a signaling molecule is methylmalonic acid (MMA), a byproduct of propionyl-CoA catabolism, which funnels metabolites from the breakdown of branched chain amino acids and lipids into the tricarboxylic acid cycle for replenishment. Vitamin B12 (cobalamin) deficiency and mutations in key propionate pathway enzymes, such as cobalamin adenosyltransferase (encoded by MMAB), lead to the accumulation of MMA. MMAB converts cobalamin to its biologically active form, which maintains flux to break down propionyl-CoA, and loss-of-function mutations lead to lethal metabolic defects and chronic liver disease in humans. Importantly, recent work identified MMAB as a key driver of a liver-specific gene regulatory co-expression network associated with altered blood lipid and glucose levels in humans, suggesting that MMAB may play a more fundamental role in coordinating appropriate physiological responses to nutrient availability. Concurrently, our group found that by modulating propionate byproducts (namely, MMA), hepatic MMAB serves as a negative feedback regulator of cholesterol biosynthesis. Our recent pilot data reveal that in humans and in preclinical mouse models of non-alcoholic fatty liver disease (NAFLD) and steatohepatitis (NASH), hepatic MMAB expression is significantly downregulated. Further, MMA has recently been associated with advanced fibrosis risk in patients with NAFLD, but substantial work is needed to define the mechanistic underpinnings of these important physiological relationships. Here, we will execute an experimental plan with two specific aims to test our central hypothesis that hepatic MMAB is a key mitochondrial metabolic checkpoint that regulates cellular phenotype and behavior. We posit that excess cholesterol disrupts this sensing network in the liver, thereby increasing MMA levels and downstream intra- and extra-hepatic signaling in ways that increase the progression of NAFLD/NASH. Here, we propose to (1) Elucidate the role of hepatic cobalamin-dependent metabolism during the progression of steatotic liver disease and (2) Define the molecular mechanisms by which hepatic MMAB contributes to the progression of NAFLD/NASH. Successful completion of the proposed studies will provide critical preliminary data for a future R01 application, as my laboratory seeks to uncover mechanism-based therapeutic target(s) to treat steatotic liver disease.
2023
Romina Bevacqua (Mt Sinai)
Innovative Genetic Technologies For Identifying Novel Regulatory Mechanisms Controlling Human b-Cell Maturation and Function.
Diabetes mellitus affects 563 million people worldwide. All forms of diabetes are consequence of impaired function or survival of pancreatic islet b cells. Importantly, hallmark functions of human pancreatic β cells improve in the years after birth. Recent studies have revealed numerous potential regulators of adult islet function, many of which are not expressed in mouse or immature human β-like cells. Understanding the genetic and molecular mechanisms regulated by these factors in adult human islets could uncover novel druggable targets for diabetes treatment and improve outcomes with β-cells from renewable sources. However, experimental approaches for genetic modeling and modulation of adult human islets are still limited.
Multiple studies have shown that transcription factors play a critical role in regulating β-cell maturation. During my postdoctoral studies, I identified novel transcriptional regulators of mature islet function, SIX2 and SIX3. I also combined primary human islet organoid systems (“pseudoislets”) with a variety of genetic approaches, including lenti-shRNAs and lenti-CRISPR, to study their roles in the adult β-cell and discover how they are regulated by non-coding mechanisms. Following a more extensive analysis, I have now identified multiple additional human transcription factors (including RXRG, NR0B1, MXI1, ONECUT2 and ESR1) that, like SIX2 and SIX3, increase their expression with age in humans. Importantly, these are unique to human beta cell/islet maturation: they are not expressed in adult mouse islet cells or β-like cells from renewable sources.
In this proposal, I will use my novel pseudoislet-genetics approach combined with lenti-shRNAs to target a subset of these recently identified candidates and explore how their depletion affects glucose-stimulated insulin secretion. Overall, the experiments proposed here should prioritize novel regulators of adult human β-cell function.
While these are important advances, further refinements in genetic manipulation of primary human islets will simplify the ‘scaling’ of these gene-targeting approaches, and will introduce novel possibilities for discovery, such as multiplexed targeting of distinct regulatory elements. Thus, in this proposal, I will adapt CRISPR/Cas9 ribonucleoprotein (RNP) electroporation methods for use in adult primary islets. I have designed proof-of-concept experiments to validate this approach, including targeting of the transcription factor PDX1, followed by experiments designed to simultaneously target two regulatory elements of the key beta cell enzyme, PCSK1. These studies will constitute the basis for future studies of epistatic interactions in primary islets.
These considerations lead to the following Specific Aims:
Specific Aim 1: Elucidate The Roles Of Novel Age-Dependent TFs in Primary Human β-Cells.
Specific Aim 2: Establish CRISPR-RNP-Mediated Gene Editing in Primary Human β-Cells.
Collectively, these studies bear the potential to significantly increase our understanding of the genetic mechanisms governing human adult β-cell function, and to expand our toolkit for genetic interrogation of human islets.
Seiya Kitamura (Einstein)
Small molecule degraders of soluble epoxide hydrolase as novel therapeutics for diabetes
Although first-generation soluble epoxide hydrolase (sEH) inhibitor entered clinical trials more than 10 years ago for the treatment of insulin resistance and diabetes, there are currently still no drugs on market targeting this promising therapeutic target for diabetes. As an alternative approach to such conventional inhibitors, the first-in-class small molecule sEH degraders were recently developed by our group that promote the degradation of sEH within cells with superior cellular efficacy compared to sEH inhibitors. The long-term goal of this application is to show the proof-of-concept that small molecule induced degradation of sEH is a promising pharmacological approach for the treatment of diabetes. The central hypothesis is that small molecule sEH degrader has higher efficacy to reduce hepatic insulin resistance and BAT regression compared to sEH inhibitors. The rationale for this project is that a determination of the preclinical therapeutic efficacy of sEH degraders in animal model of diabetes will offer a strong scientific framework whereby new strategies to diabetes therapy in patients can be developed. The central hypothesis will be tested by pursuing two specific aims: (1) Test the in vivo efficacy of sEH degrader on hepatic insulin resistance, and (2) Compare the effect of sEH degrader vs inhibitor on BAT regression. Under the first aim, high fat diet-induced diabetic mouse model will be used to evaluate the in vivo efficacy of sEH degrader vs sEH inhibitor on insulin resistance of liver. For the second aim, the in vivo effects of sEH degrader on BAT regression will be evaluated in comparison with sEH inhibitor. The research proposed in this application is innovative because it focuses on sEH degrader as a new modality of sEH targeting therapeutics that show high degradation potency with prolonged and superior efficacy. The proposed project is significant because it is expected to provide strong scientific justification for the continued development and future clinical trials of sEH degraders. More in general, our proposed project will show the in vivo efficacy of the first small molecule degraders targeting diabetes, ultimately providing new opportunities for the development of novel therapeutic modality to treat diabetes.
Farnaz Shamsi (Cornell)
Targeting Adipose Tissue Neurovasculature to Improve Metabolism
Obesity affects 19% of children and 42% of adults in the US and increases the risk for the leading causes of death in the US and worldwide, including diabetes, heart disease, stroke, and some types of cancer. Brown adipose tissue (BAT) is a specialized type of adipose that is primarily responsible for regulating body temperature. Once activated by cold, BAT dissipates the chemical energy as heat in a process called adaptive thermogenesis. BAT is functionally distinct from white adipose tissue (WAT) which is the primary site of energy storage. Due to their high metabolic activity, thermogenic adipocytes act as a metabolic sink to improve glucose and lipid metabolism and thus exhibit anti-diabetic and lipid-lowering effects. The major challenge in targeting BAT thermogenesis as an anti-diabetes and anti-obesity therapy is the limited amount of active BAT in most adult humans. A critical barrier to harnessing the potential of BAT to enhance cardiometabolic health in humans is the lack of understanding of the full range of pathways involved in the activation of BAT thermogenesis. We have recently identified the axon guidance ligand Slit3 as an essential regulator of BAT thermogenesis. We showed that loss of Slit3 dramatically reduces angiogenesis and sympathetic innervation in BAT and impairs cold-induced BAT thermogenesis. However, the role of Slit3 in the establishment of the neurovascular network in developing adipose tissue and in response to obesogenic challenges is unknown. In this proposal, we will use a series of innovative strategies to determine the role of Slit3 in the early development of adipose tissue neurovasculature (Aim 1) and obesity-induced expansion and remodeling of adipose tissue (Aim 2). Since enhanced angiogenesis and sympathetic innervation in adipose tissue is linked to improved metabolic health, we will address the potential of Slit3 to ameliorate the detrimental effects of diet-induced and genetic obesity by promoting angiogenesis and sympathetic innervation in BAT and WAT. Successful completion of the proposed studies will identify a new potential node of intervention for obesity and metabolic diseases by stimulating the healthy expansion of adipose tissue.
Linheng Wang (Mt Sinai)
Mechanisms of TOX4 deficiency-induced steatosis
The liver plays a central role in systemic glucose and lipid homeostasis. Nonalcoholic fatty liver disease (NAFLD) is the most common liver disease and is strongly associated with obesity (70-90%) and T2D (80%). NAFLD is driven by complex inherited and environmental factors related to obesity, insulin resistance, and dyslipidemia. Currently, there is no approved drug for treating NAFLD due to a limited understanding of its pathological mechanisms. The PI has recently identified TOX High Mobility Group Box Family Member 4 (TOX4) as a novel insulin-regulated TF that regulates hepatic glucose production in parallel with the classic insulin receptor/FoxO1 pathway. Interestingly, acute knockdown or congenital knockout of TOX4 in hepatocytes robustly promotes lipid accumulation in the liver. Conversely, overexpression of TOX4 in primary hepatocytes suppresses lipogenesis and reduces liver lipid accumulation in vivo. Further RNA-seq analysis of liver samples from TOX4 flox and liver-specific knockout (TLKO) mice revealed that the top differentially expressed genes are enriched in lipid metabolic pathways, suggesting TOX4 as a novel negative regulator of lipid synthesis. Furthermore, complement factor D (CFD, also named adipsin) is among the top upregulated genes in the TLKO liver. Indeed, CFD is elevated in the livers of human NAFLD subjects and obese mice, distinct from its circulating levels and adipose expression. Whole-body knockout of CFD protects mice from liver steatosis. Therefore, the PI hypothesizes that hepatocyte TOX4 protects from NAFLD through transcriptional repression of lipogenic genes and CFD. In Aim 1, the PI will first perform chromatin immunoprecipitation sequencing (ChIP-seq) to determine the genome-wide landscape of TOX4-mediated transcription regulation under normal and pathological conditions, focusing on the loci of lipogenic genes. Combining ChIP-seq and RNA-seq analyses, she will identify the direct and indirect target genes of TOX4, which will provide insights into subsequent mechanistic studies. In Aim 2, she will first establish CFD as a downstream target of TOX4 and begin to elucidate the repressive mechanisms. She will generate mice to specifically ablate CFD in TLKO hepatocytes and critically test whether TOX4 deficiency promotes steatosis through a CFD-dependent mechanism. Successful completion of the proposed studies in this application will illustrate the function and mechanism of TOX4 in regulating hepatic lipid metabolism. This proposal will identify novel genes that are critical for the development of NAFLD and therefore serve as therapeutic targets. The proposal will also help the PI to generate essential preliminary data to apply for the R01 grant.
2022
Simone Sidoli, PhD, Einstein
Deciphering the role of the metabolically-induced histone modification H3K27ac in Heart Failure with Preserved Ejection Fraction (HFpEF) syndrome in patient samples and a new mouse model
Project Summary/Abstract:
There is a fundamental gap in understanding the bi-directional mechanisms by which perturbed metabolic states such as that in diabetes, influence epigenetic mechanisms to drive pathologic gene expression. The Heart Failure with Preserved Ejection Fraction (HFpEF) syndrome, widely considered the greatest unmet therapeutic need in cardiology, has rapidly become the most prevalent form of heart failure due to its intimate relationship with diabetes and metabolic syndrome. Multi-omic studies from our group have helped to establish transcriptomic, proteomic, and metabolomic phenotypes of modern HFpEF, notably one associated with diabetes and metabolic syndrome (HFpEF-diabetes). This makes HFpEF-diabetes the ideal stage to study metaboloepigenetic mechanisms by which diabetes drives multi-organ dysfunction. Recent studies show that metabolic perturbations can influence relative abundances of histone post-translational modifications (PTMs), aka “histone marks”, and thus DNA readout. The long-term goal of my proposed research is to identify how metabolic dysfunction drives disease pathology via altering chromatin state, with the initial objective to define histone PTM signatures in HFpEF-diabetes and to uncover the mechanisms by which they drive myocardial dysfunction in HFpEF-diabetes. The central hypothesis is that diabetes alters histone PTMs such as H3K27ac, driving aberrant gene expression programs that regulate myocardial metabolism, worsening HFpEF. Guided by strong preliminary data, this hypothesis will be tested by pursuing two specific aims: 1) Elucidate the mechanisms by which H3K27ac and other histone marks modulate gene expression programs in endomyocardial biopsies from HFpEF-diabetes patients; and 2) Determine the signaling pathways by which H3K27ac and other marks mediate metabolic and diastolic dysfunction in laboratory models of HFpEF-diabetes. Under the first aim, the applicant’s unique access to endomyocardial biopsy tissue obtained in collaboration with the Center For Heart Failure With Preserved Ejection Fraction clinic at the Johns Hopkins Hospital, will be coupled with the applicant’s pioneering expertise to measure histone PTMs globally using mass spectrometry. Under the second aim, relevant animal models of HFpEF-diabetes will be identified to determine the most relevant models for mechanistic studies. This approach is innovative because it couples the applicant’s unique expertise in measuring histone PTMs in microgram-sized amounts of endomyocardial biopsy tissue, with multi-omic analyses to uncover relationships between clinical variables and biologic mechanisms. The proposed research is significant because it is expected to vertically advance and expand understanding of how perturbed metabolic states in diabetes drive multi-organ dysfunction via metaboloepigenetic mechanisms. Ultimately, such knowledge has the potential to better phenotype HFpEF-diabetes patients, and to identify novel epigenetic and metabolic therapies.
Lauryn Choleva, MD, Mt. Sinai
Structure-Function Studies of p57KIP2 in The Human Pancreatic Beta Cell
Abstract
I am an Instructor in the Department of Pediatric Endocrinology at the Icahn School of Medicine at Mount Sinai. During my clinical fellowship in Pediatric Endocrinology and Diabetes, I spent part of my time developing research skills in the laboratory of Dr. Andrew Stewart in Mount Sinai’s Diabetes Obesity Metabolism Institute (DOMI). As a new Instructor, I spend 1 full day in clinic and the remaining 80% of my time devoted to continuing basic research.
My work focuses on the cell cycle inhibitor, p57KIP2, encoded by the CDKN1C gene on chromosome 11. p57KIP2 is of particular interest in pediatric endocrinology, since mutations in CDKN1C underlie multiple pediatric endocrine syndromes. These include Beckwith-Wiedemann Syndrome (BWS), IMAGe Syndrome, Russel-Silver Syndrome (RSS), and the Focal Variant of Congenital Hyperinsulinism (FoCHI). In addition, p57KIP2 is reduced in insulinomas in adults. Finally, human β cell regenerative drugs lead to suppression of p57KIP2. Thus, p57KIP2 is central both to rare pediatric endocrine syndromes, as well as to therapies for Type 1 and Type 2 Diabetes.
Surprisingly, despite its importance, p57KIP2 is very poorly studied. p57KIP2 is a member of the CIP-KIP family of cell cycle inhibitors that also includes p21CIP1 and p27KIP1. In contrast to other cell cycle inhibitors, it is distinguished by the presence of a large proline-alanine-rich domain, and by its expression in only a subset of beta cells. Remarkably, unlike other cell cycle inhibitors, p57KIP2 is expressed in most human tissues during embryogenesis, but is limited in adult humans to only a few tissues, one of which is the human beta cell. And finally, the CDKN1C locus is maternally imprinted. Remarkably, despite this central role in pediatric disease and beta cell biology, very little is known about the function of p57KIP2 in any cell type, including the human beta cell. I hypothesize that p57KIP2 has yet-unknown functions that extend beyond cell cycle control and are unique to the human beta cell. In this proposal, I will perform the first structure-function studies of human p57KIP in the human beta cell. These studies comprise the first complete detailed analysis of this important, but understudied, human disease-associated gene and protein.
Jessica Ables, PhD, Mt. Sinai
Regulation of Extracellular Matrix by Chronic Hyperglycemia May Contribute to Behavioral Dysregulation in Diabetes
Patients with diabetes are at risk for neurocognitive deficits, including depression and anxiety. Examination of brains from people with Type 2 diabetes indicates that the majority of gene expression changes are found in the reward circuitry, suggesting that diabetes may affect reward circuitry function to contribute to the increased incidence of mood disorders. To explore this possibility, I have examined gene expression in cholinergic medial habenula (MHb) neurons, a brain area that is associated with anhedonia when damaged, and found upregulation of 176 genes, including genes involved in neurotransmission, and cell-cell adhesion after 12 weeks of hyperglycemia in STZ-treated mice. A much larger number of genes were downregulated in the MHb (1075), including a significant fraction involved in extracellular matrix (ECM) remodeling. For example, matrix-metalloproteinase 2 (MMP2) is downregulated in several brain areas in human patients with depression and is also downregulated in MHb neurons of diabetic mice. Here we propose to first characterize the ECM of reward circuitry and its dysregulation in 3 mouse models of diabetes (db/db, high fat diet and STZ). The second aim will examine the behavioral implications of manipulating ECM in the MHb. Together these aims will provide evidence for one mechanism by which chronic hyperglycemia can increase the risk for mood disorders.
2021
Sharon Rikin, MD, Einstein
Multidisciplinary electronic consultation to improve evidence-based care for patients with diabetic kidney disease
The number of patients with diabetic kidney disease (DKD) is increasing with existing health care delivery models failing to implement evidence-based guidelines that can prevent progression to end stage kidney disease (ESKD). The overarching goal of this pilot and feasibility proposal is to develop and pilot-test a Multidisciplinary Diabetic Kidney Disease (DKD) Program, to promote primary care provider (PCP) prescribing of SGLT2i and RAASi for patients at high risk of progression from DKD to ESKD. It is well known that patients with diabetes experience gaps in evidence-based treatment. Barriers to evidence-based treatment include complex but modifiable physician and health-system related barriers such as competing preventive and chronic disease priorities, discomfort with new medications, and lack of multidisciplinary collaboration for a disease at the intersection between primary care, endocrinology and nephrology. Developing effective interventions to improve the use of evidence-based treatments to prevent the progression of DKD should be a national priority.
The proposed Multidisciplinary Diabetic Kidney Disease Program consists of three elements: 1) multidisciplinary collaboration between endocrinologists, nephrologists and PCPs, 2) an electronic consultation sent from endocrinologists and nephrologists to PCPs which includes decision support to address care gaps customized for individual patients with high risk DKD, 3) a registry of patients with high risk DKD which will enable proactive targeting of patients with gaps in evidence-based treatment. With user-centered design principles, we will conduct focus groups with stakeholders from primary care, endocrinology, and nephrology to rigorously develop and iteratively refine the intervention in order to overcome identified barriers to guideline-driven care. We will then pilot-test the intervention in a randomized controlled trial. We will use mixed qualitative and quantitative methods to assess the feasibility, acceptability and preliminary effectiveness of the Multidisciplinary Diabetic Kidney Disease Program.
We hypothesize that primary care physicians randomized to receive the electronic consult intervention, will find the intervention acceptable, appropriate and feasible. We also hypothesize that patients whose physician received the intervention, compared to no intervention, will be more likely to be prescribed SGLTi and RAASi at maximum dose. This study will lay the foundation for extramural funding of a future large randomized controlled trial to test the effectiveness of the intervention to reduce DKD progression in those at high risk. Study results have the potential to influence redesign of health care delivery for diabetes care and may serve as a model of clinical decision support for these and other high yield evidence-based guidelines.
Hongling Zhao, PhD, Einstein
Targeting Skp2/Cks1-p27 pathway in the treatment of obesity induced by loss of Rb1 in POMC neurons and high fat diet.
The Arcuate nucleus (ARC) in hypothalamus contains antagonizing POMC and AGRP/NPY neurons for negative and positive energy balance, respectively. We have examined the role of cell cycle regulation in POMC and AGRP/NPY neurons as a means of discovering a molecular mechanism for dysfunction of POMC neurons in animals fed a high fat diet. We found that the maintenance of POMC neurons in a quiescent cell cycle phase is an active process, requiring the activity of a normal Retinoblastoma (pRb) protein to prevent neuronal dysfunction. High fat feeding deactivates pRb by inducing pRb phosphorylation and E2F target gene de‐repression in POMC neurons by cyclin dependent kinase 4 (CDK4) and the resulting obesity can be prevented by treatment with a CDK4 inhibitor, abemaciclib. Our findings repurpose this class of breast cancer treatment drugs could be a new modality for treatment of obesity. Skp2 is an E3 ubiquitin ligase and responsible for substrate recognition in SCF ubiquitin ligase complex. The best-established function of the SCFSkp2 ubiquitin ligase is to mediate ubiquitination of p27, which prepares p27 for degradation in the proteasome. By interacting with the N terminus of Skp2, Rb protein interferes with Skp2-p27 interaction, and inhibits ubiquitination of p27. p27, which is also called cyclin-dependent kinase inhibitor 1B (CDKN1B), is a potent inhibitor of cyclin-CDK complexes (cyclin E-CDK2 and cyclin D-CDK4) and controls cell cycle progression at G1. Our preliminary data strongly support the hypothesis that codeletion of pRb target Skp2 might also prevent obesity induced by loss of Rb1 in POMC neurons and high fat feeding. Since Skp2-mediated p27 proteasome degradation also requires an accessory protein Cks1 (to form SCFSkp2/Cks1), we believe that Cks1 mutant, N45R, which prevents Skp2-Cks1 binding and subsequent p27 proteasome degradation, also inhibits the manifestation of the full obesity phenotype associated with loss of Rb1 in POMC neurons and high fat feeding. In this project, we are going to take advantage of two novel mouse models, Cks1 N45R knockin mouse model and conditional Skp2 knockout mouse model, to dissect the underlying mechanisms of preventive and therapeutic effects of targeting Skp2/Cks1-p27 on obesity induced by loss of Rb1 in POMC neurons and high fat feeding. We purpose that Skp2/Cks1 inhibitors could be a new modality for treatment of obesity, which might provide basis for applying another class of cancer treatment drugs to obesity treatment.
We propose the following Specific Aims to test this hypothesis:
Aim 1. To determine the mechanisms of the preventive effect of Skp2KO and Cks1 N45R KI on obesity induced by loss of Rb1 in POMC neurons and high fat feeding.
Aim 2. To determine whether the preventive effect of Skp2KO on obesity induced by loss of Rb1 in POMC neurons and high fat feeding is cell autonomous.
Aim 3. To determine the impact of Skp2 inhibition on hypothalamic POMC neurons and obesity.
2020
Esra Karakose PhD (Mt. Sinai)
Mechanisms of Epigenetic Regulation of Human Pancreatic Beta Cell Replication
Diabetes results from loss or deficiency of insulin-producing beta cells. Restoring normal beta cell mass and function is the ultimate goal to reverse both Type 1 and Type 2 diabetes. Although various approaches to replenish beta cells, such as pancreas or islet transplantation and stem cell-based beta cell differentiation strategies, are in various stages of development or limited clinical use, many challenges remain before they can enter widespread clinical use. Since there is a residual beta cell pool in most patients with diabetes, an alternate promising approach would be to discover drugs that are able to induce the replication of the residual beta cells to restore beta cell mass to normal.
In our previous studies, we showed that a rare and benign type of pancreatic neuroendocrine tumor called “insulinoma” holds the “genomic recipe” for the molecular pathways that induce human beta cell replication. Further, we demonstrated that epigenetic regulation is key to control molecular mechanisms of beta cell replication. The studies in this proposal will address the gap of knowledge in epigenetic regulation of beta cell replication. We will decipher how exactly the epigenetic alterations impact the beta cell replication at a molecular level allowing us to discover new drug targets and druggable pathways. First, we will build a statistically validated normative human beta cell ATAC-seq and chromatin mark ChIP-seq database. Second, we will compare the open chromatin landscape as well as the loci that harbor active and repressive histone marks in insulinomas vs. beta cells. Third, we will compare open/closed chromatin regions and active and repressive histone mark alterations in benign insulinomas to those in malignant and nonfunctional PNETs, to discern pathways involved in benign, as opposed to malignant, beta cell proliferation.
Angela Lombardi, PhD (Einstein)
Innovative therapeutic strategies in Type 1 Diabetes
We hypothesize that the presentation of pathogenic pancreatic peptides to T-cells within the HLA-DQ8 pocket is a key trigger in T1D, and that blocking the peptide binding to this pocket can be harnessed to treat/prevent the autoimmune response targeting beta cells in T1D. We will perform ex vivo and in vivo experiments using transgenic humanized DQ8-mice and DQ8-peripheral blood mononuclear cells(PBMCs) isolated from T1D patients, which provide exquisite tools to test our hypothesis. Since T1D results from killing and/or silencing beta cells by various immune effectors mechanisms, several non-targeted immune therapies have been proposed in order to block the immune attack. However, these therapies have unacceptable toxicity, making novel therapies urgently needed. The goal of the present study was to test an innovative peptide therapeutics approach to treat T1D by blocking the interaction between HLA-DQ8 positive antigen presenting cells (APCs)and CD4+ T-cells. Peptide therapeutics provide greater efficacy, selectivity, specificity, and safety compared to small molecules and antibodies. To overcome the peptides’ intrinsic disadvantage of sensitivity to proteases we harnessed D-amino acid based peptides harboring the reverse sequence (retro-inverso) of the corresponding L-amino acid peptides (D-peptides).We provide strong preliminary data indicating that we have 2 potential candidates able to suppress T-cell activation by blocking the presentation of InsB:9-23 peptide within HLA-DQ8 pocket. Indeed, we identified two D-peptides that inhibit InsB:9-23 binding to recombinant HLA-DQ8 molecule, as well as its binding to DQ8 expressed on human B-cells. Moreover, both peptides prevent T-cell activation in a cellular antigen presentation assay containing human DQ8 cells loaded with InsB:9-23 peptide and murine T-cells expressing a human TCR specific for the InsB:9-23–DQ8 complex. On these grounds, we will explore the following specific aims: in Aim 1we will test HLA-DQ8-InsB:9-23 D-peptides blockers ex vivo in humanized HLA-DQ8 mice and in human PBMCs isolated from T1D patients; whereas Aim 2 will analyze the effect of the D-peptides in vivo in DQ8 mice and in NOD mice.
Prashant Rajbhandari, PhD (Mt Sinai)
Transcriptional control of adipogenesis by transcription factor PATZ1
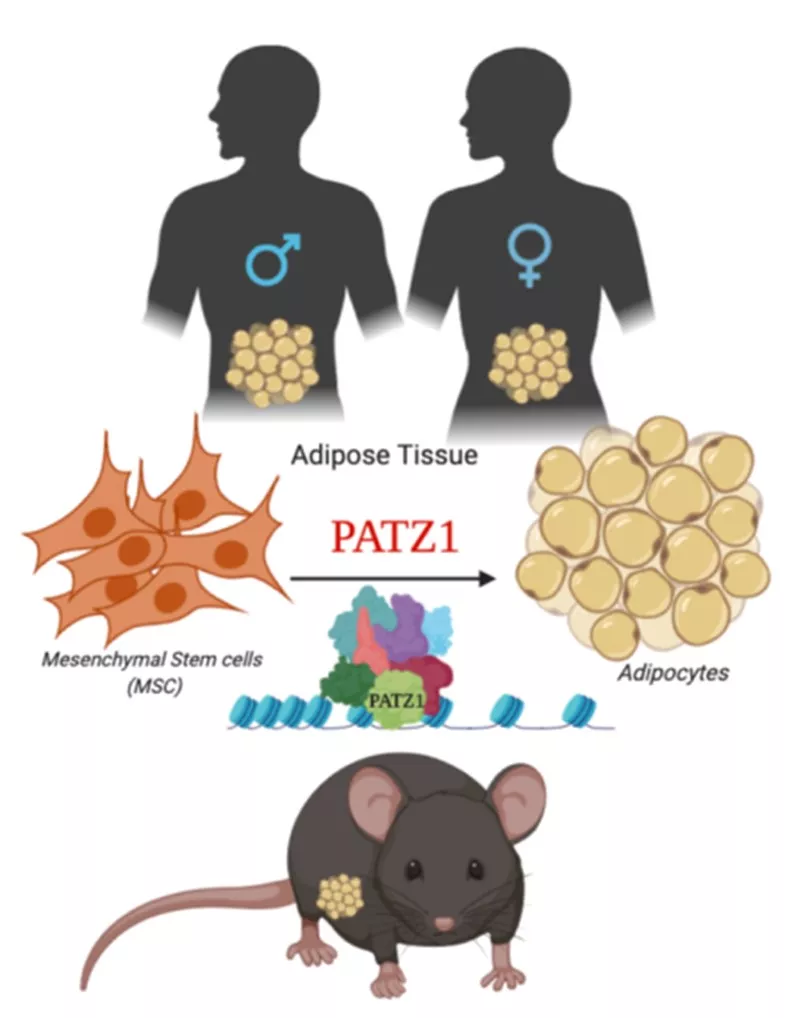
Video could not be played
Fig. 1. Model depicting proposed role of PATZ1 as a transcriptional modulator of adipogenesis in mice and humans.
Adipocytes are remarkably complex endocrine cells that regulate energy homeostasis and understanding the intricacies of the processes of adipocyte formation is a major relevance to obesity and Type 2 diabetes. We have identified zinc finger protein, POZ/BTB and AT Hook Containing Zinc Finger 1 (PATZ1) as a novel adipogenic transcription factor through an unbiased high-throughput cDNA screen for modulators of adipogenesis. PATZ1 expression correlates with murine obesity, and in vitro adipocyte differentiation studies confirm the ability of PATZ1 to promote adipogenesis through a mechanism dependent on DNA binding. Global analyses using ChIP-Seq suggest that PATZ1 facilitates adipogenesis through interactions with transcription factor machinery at the start sites of key adipogenic factors and histone modifiers. Adipocyte-specific ablation of PATZ1 in mice leads to decreasedfat mass and protection from diet-induced obesity in a sex-specific manner. To further test the hypothesis that adipose-specific PATZ1 imprints fate of mesenchymal stem cells and preadipocytes (Fig. 1) and determines a rewiring of their metabolic capacity, I will aim to i) elucidate the role of PATZ1 in adipose physiology and metabolism in males and females and ii) determine the molecular mechanism by which PATZ1 controls adipogenesis. The findings from the proposal will further our understanding of the complex transcriptional mechanisms underlying adipose tissue homeostasis and systemic energy balance.